

Description
Beta-propeller protein-associated neurodegeneration (BPAN) is a disorder that damages the nervous system and is progressive, which means that it gradually gets worse. Affected individuals develop a buildup of iron in the brain that can be seen with medical imaging. For this reason, BPAN is classified as a type of disorder called neurodegeneration with brain iron accumulation (NBIA), although the iron accumulation may not occur until late in the disease.
Many people with BPAN have recurrent seizures (epilepsy) beginning in infancy or early childhood. Several different types of seizures can occur in this disorder, even in the same individual. Often the first type to occur are febrile seizures, which are triggered by a high fever. Affected individuals can also experience generalized tonic-clonic seizures (also known as grand mal seizures). This type of seizure affects the entire body, causing muscle rigidity, convulsions, and loss of consciousness. Other seizure types that can occur in this disorder include short lapses in awareness that can have the appearance of staring spells or daydreaming (absence seizures, also called petit mal seizures), sudden episodes of weak muscle tone (atonic seizures), involuntary muscle twitches (myoclonic seizures), or more pronounced movements called epileptic spasms. Some individuals have seizure patterns that resemble those in epileptic syndromes, such as West syndrome or Lennox-Gastaut syndrome.
can occur in this disorder, even in the same individual. Often the first type to occur are febrile seizures, which are triggered by a high fever. Affected individuals can also experience generalized tonic-clonic seizures (also known as grand mal seizures). This type of seizure affects the entire body, causing muscle rigidity, convulsions, and loss of consciousness. Other seizure types that can occur in this disorder include short lapses in awareness that can have the appearance of staring spells or daydreaming (absence seizures, also called petit mal seizures), sudden episodes of weak muscle tone (atonic seizures), involuntary muscle twitches (myoclonic seizures), or more pronounced movements called epileptic spasms. Some individuals have seizure patterns that resemble those in epileptic syndromes, such as West syndrome or Lennox-Gastaut syndrome.
Children with BPAN also have intellectual disability, delayed development including significant problems with vocabulary and producing speech (expressive language), and difficulty coordinating movements (ataxia). Ataxia can affect the ability to walk and perform fine motor skills such as using utensils. Affected individuals can have neurodevelopmental issues that are often compared to features of a disorder called Rett syndrome. These features include repeated hand wringing or clasping (stereotypic hand movements); teeth grinding (bruxism); sleep disturbances; and problems with communication and social interaction characteristic of autism spectrum disorder.
In late adolescence or early adulthood, individuals with BPAN may begin to experience a gradual loss of intellectual functioning (cognitive decline) that can lead to a severe loss of thinking and reasoning abilities (dementia). Worsening problems with movement also occur, including dystonia and parkinsonism. Dystonia is a condition characterized by involuntary, sustained muscle contractions. In BPAN, the dystonia often starts in the arms. Parkinsonism can include unusually slow movement (bradykinesia), rigidity, tremors, an inability to hold the body upright and balanced (postural instability), and a shuffling walk that can cause recurrent falls.
The lifespan of people with BPAN varies. With proper management of their signs and symptoms, affected individuals can live into middle age. Death may result from complications of dementia or movement problems, such as injuries from falls or swallowing difficulties (dysphagia) that can lead to a bacterial lung infection called aspiration pneumonia.
Frequency
BPAN is a rare disorder. Its prevalence is unknown, but it is thought to account for between 35 and 40 percent of all cases of NBIA disorders. Some individuals who have been diagnosed with intellectual disability or early-onset parkinsonism based on their signs and symptoms have later been found to have BPAN when genetic testing was done.
Causes
BPAN is caused by variants (also called mutations) in the WDR45 gene. This gene provides instructions for making the WIPI4 protein. WIPI4 has a characteristic structure resembling a seven-bladed propeller, from which the name of the disorder is derived. The WIPI4 protein is involved in a process called autophagy, which helps clear unneeded materials from cells, including excess amounts of an iron storage protein called ferritin .
.
Most of the WDR45 gene variants identified in people with BPAN result in the absence of functioning WIPI4 protein. As a result, the process of autophagy is impaired, making cells less efficient at removing damaged cell structures and waste materials. Researchers suggest that nerve cells (neurons ) may be particularly vulnerable to impaired autophagy because they have long extensions (axons and dendrites), making it even more difficult to transport the waste materials from these structures to the compartments in the cell body where recycling takes place (the lysosomes
) may be particularly vulnerable to impaired autophagy because they have long extensions (axons and dendrites), making it even more difficult to transport the waste materials from these structures to the compartments in the cell body where recycling takes place (the lysosomes ). The waste materials can build up in these areas and damage them. Damage to neurons results in the neurological problems that occur in BPAN.
). The waste materials can build up in these areas and damage them. Damage to neurons results in the neurological problems that occur in BPAN.
Inheritance
This condition is inherited in an X-linked dominant pattern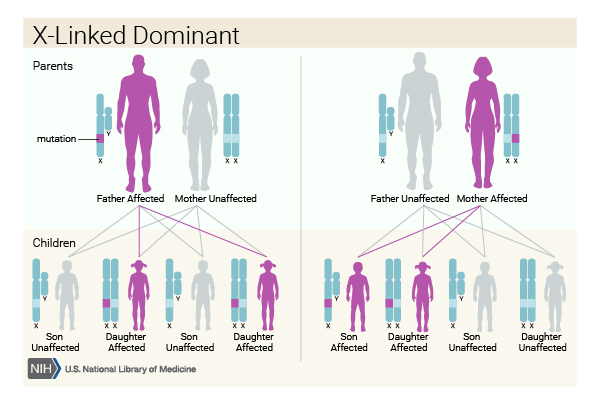 . The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes
. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes . In females (who have two X chromosomes), a variant in one of the two copies of the gene in each cell is sufficient to cause the disorder. In males (who have only one X chromosome), a variant in the only copy of the gene in each cell causes the disorder. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
. In females (who have two X chromosomes), a variant in one of the two copies of the gene in each cell is sufficient to cause the disorder. In males (who have only one X chromosome), a variant in the only copy of the gene in each cell causes the disorder. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
In most X-linked dominant disorders, males experience more severe symptoms than females. While this is not always the case in BPAN, most individuals with the disorder are females, likely because a smaller number of affected males survive until birth.
Almost all cases of BPAN result from new variants in the gene and occur in people with no history of the disorder in their family. Rarely, an affected person inherits the variant from a mildly affected mother. Among reported cases, males with BPAN and most females with BPAN have not had children.
Other Names for This Condition
- BPAN
- NBIA5
- Neurodegeneration with brain iron accumulation 5
- SENDA
- Static encephalopathy of childhood with neurodegeneration in adulthood
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Ebrahimi-Fakhari D, Saffari A, Wahlster L, Lu J, Byrne S, Hoffmann GF, Jungbluth H, Sahin M. Congenital disorders of autophagy: an emerging novel class of inborn errors of neuro-metabolism. Brain. 2016 Feb;139(Pt 2):317-37. doi: 10.1093/brain/awv371. Epub 2015 Dec 29. Citation on PubMed
- Gregory A, Kurian MA, Haack T, Hayflick SJ, Hogarth P. Beta-Propeller Protein-Associated Neurodegeneration. 2017 Feb 16. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK424403/ Citation on PubMed
- Haack TB, Hogarth P, Gregory A, Prokisch H, Hayflick SJ. BPAN: the only X-linked dominant NBIA disorder. Int Rev Neurobiol. 2013;110:85-90. doi: 10.1016/B978-0-12-410502-7.00005-3. Citation on PubMed
- Hayflick SJ, Kruer MC, Gregory A, Haack TB, Kurian MA, Houlden HH, Anderson J, Boddaert N, Sanford L, Harik SI, Dandu VH, Nardocci N, Zorzi G, Dunaway T, Tarnopolsky M, Skinner S, Holden KR, Frucht S, Hanspal E, Schrander-Stumpel C, Mignot C, Heron D, Saunders DE, Kaminska M, Lin JP, Lascelles K, Cuno SM, Meyer E, Garavaglia B, Bhatia K, de Silva R, Crisp S, Lunt P, Carey M, Hardy J, Meitinger T, Prokisch H, Hogarth P. beta-Propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain. 2013 Jun;136(Pt 6):1708-17. doi: 10.1093/brain/awt095. Epub 2013 May 17. Citation on PubMed or Free article on PubMed Central
- Nishioka K, Oyama G, Yoshino H, Li Y, Matsushima T, Takeuchi C, Mochizuki Y, Mori-Yoshimura M, Murata M, Yamasita C, Nakamura N, Konishi Y, Ohi K, Ichikawa K, Terada T, Obi T, Funayama M, Saiki S, Hattori N. High frequency of beta-propeller protein-associated neurodegeneration (BPAN) among patients with intellectual disability and young-onset parkinsonism. Neurobiol Aging. 2015 May;36(5):2004.e9-2004.e15. doi: 10.1016/j.neurobiolaging.2015.01.020. Epub 2015 Jan 30. Citation on PubMed
- Saitsu H, Nishimura T, Muramatsu K, Kodera H, Kumada S, Sugai K, Kasai-Yoshida E, Sawaura N, Nishida H, Hoshino A, Ryujin F, Yoshioka S, Nishiyama K, Kondo Y, Tsurusaki Y, Nakashima M, Miyake N, Arakawa H, Kato M, Mizushima N, Matsumoto N. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet. 2013 Apr;45(4):445-9, 449e1. doi: 10.1038/ng.2562. Epub 2013 Feb 24. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.