

Description
Joubert syndrome is a disorder that affects many parts of the body. The signs and symptoms of this condition vary among affected individuals, even among members of the same family.
The hallmark feature of Joubert syndrome is a combination of brain abnormalities that together are known as the molar tooth sign, which can be seen on brain imaging studies such as magnetic resonance imaging (MRI). This sign results from the abnormal development of structures near the back of the brain, including the cerebellar vermis and the brainstem. The molar tooth sign got its name because the characteristic brain abnormalities resemble the cross-section of a molar tooth when seen on an MRI.
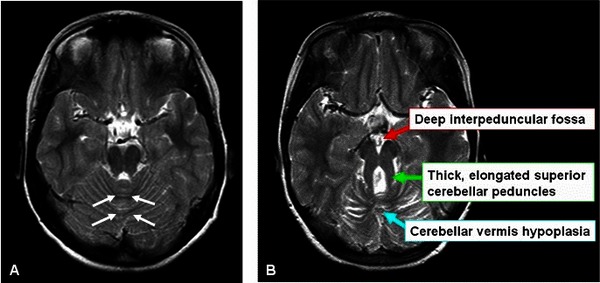
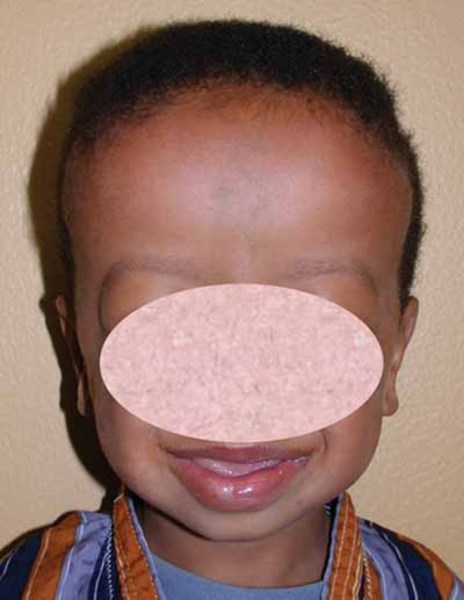
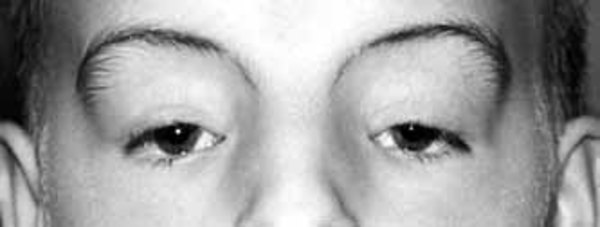
Most infants with Joubert syndrome have low muscle tone (hypotonia) in infancy, which contributes to difficulty coordinating movements (ataxia) in early childhood. Other characteristic features of the condition include episodes of unusually fast (hyperpnea) or slow (apnea) breathing in infancy, and abnormal eye movements (ocular motor apraxia). Most affected individuals have delayed development and intellectual disability, which can range from mild to severe. Distinctive facial features can also occur in Joubert syndrome; these include a broad forehead, arched eyebrows, droopy eyelids (ptosis), widely spaced eyes (hypertelorism), low-set ears, and a triangle-shaped mouth.
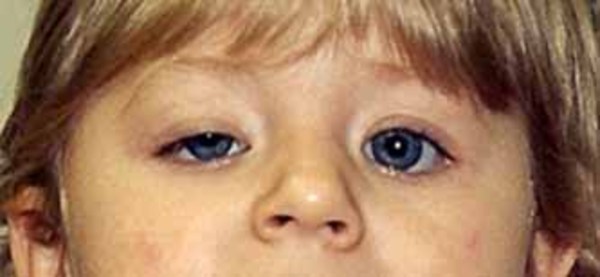

Joubert syndrome can include a broad range of additional signs and symptoms. The condition is sometimes associated with other eye abnormalities (such as retinal dystrophy, which can cause vision loss, and coloboma, which is a gap or split in a structure of the eye), kidney disease (including polycystic kidney disease and nephronophthisis), liver disease, skeletal abnormalities (such as the presence of extra fingers and toes), or hormone (endocrine) problems. A combination of the characteristic features of Joubert syndrome and one or more of these additional signs and symptoms once characterized several separate disorders. Together, those disorders were referred to as Joubert syndrome and related disorders (JSRD). Now, however, any instances that involve the molar tooth sign, including those with these additional signs and symptoms, are usually considered Joubert syndrome.
Frequency
Joubert syndrome is estimated to affect between 1 in 80,000 and 1 in 100,000 newborns. However, this estimate may be too low because Joubert syndrome has such a large range of possible features and is likely underdiagnosed. Particular genetic mutations that cause this condition are more common in certain ethnic groups, such as Ashkenazi Jewish, French-Canadian, and Hutterite populations.
Causes
Joubert syndrome can be caused by mutations in more than 30 genes. The proteins produced from these genes are known or suspected to play roles in cell structures called primary cilia. Primary cilia are microscopic, finger-like projections that stick out from the surface of cells and are involved in sensing the physical environment and in chemical signaling. Primary cilia are important for the structure and function of many types of cells, including brain cells (neurons) and certain cells in the kidneys and liver. Primary cilia are also necessary for the perception of sensory input, which is interpreted by the brain for sight, hearing, and smell.
Mutations in the genes associated with Joubert syndrome lead to problems with the structure and function of primary cilia. Defects in these cell structures can disrupt important chemical signaling pathways during development. Although researchers believe that defective primary cilia are responsible for most of the features of these disorders, it is not completely understood how they lead to specific developmental abnormalities.
Mutations in the genes known to be associated with Joubert syndrome account for about 60 to 90 percent of all cases of this condition. In the remaining cases, the genetic cause is unknown.
Inheritance
Joubert syndrome typically has an autosomal recessive pattern of inheritance, which means both copies of a gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they usually do not show signs and symptoms of the condition.

Rare cases of Joubert syndrome are inherited in an X-linked recessive pattern. In these cases, the causative gene is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.


Other Names for This Condition
- Agenesis of cerebellar vermis
- Cerebello-oculo-renal syndrome
- Cerebellooculorenal syndrome 1
- CORS
- Familial aplasia of the vermis
- JBTS
- Joubert-Bolthauser syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- JOUBERT SYNDROME 1; JBTS1
- JOUBERT SYNDROME 10; JBTS10
- JOUBERT SYNDROME 2; JBTS2
- JOUBERT SYNDROME 3; JBTS3
- JOUBERT SYNDROME 5; JBTS5
- JOUBERT SYNDROME 6; JBTS6
- JOUBERT SYNDROME 9; JBTS9
- JOUBERT SYNDROME 8; JBTS8
- JOUBERT SYNDROME 4; JBTS4
- JOUBERT SYNDROME 7; JBTS7
- JOUBERT SYNDROME 23; JBTS23
- JOUBERT SYNDROME 20; JBTS20
- JOUBERT SYNDROME 17; JBTS17
- JOUBERT SYNDROME 21; JBTS21
- JOUBERT SYNDROME 24; JBTS24
- JOUBERT SYNDROME 15; JBTS15
- JOUBERT SYNDROME 16; JBTS16
- JOUBERT SYNDROME 27; JBTS27
- JOUBERT SYNDROME 28; JBTS28
- JOUBERT SYNDROME 13; JBTS13
- JOUBERT SYNDROME 25; JBTS25
- JOUBERT SYNDROME 26; JBTS26
- JOUBERT SYNDROME 22; JBTS22
- JOUBERT SYNDROME 18; JBTS18
- NEPHRONOPHTHISIS 14; NPHP14
- JOUBERT SYNDROME 14; JBTS14
Scientific Articles on PubMed
References
- Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis. 2010 Jul 8;5:20. doi: 10.1186/1750-1172-5-20. Citation on PubMed or Free article on PubMed Central
- Doherty D. Joubert syndrome: insights into brain development, cilium biology, and complex disease. Semin Pediatr Neurol. 2009 Sep;16(3):143-54. doi: 10.1016/j.spen.2009.06.002. Citation on PubMed or Free article on PubMed Central
- Maria BL, Quisling RG, Rosainz LC, Yachnis AT, Gitten J, Dede D, Fennell E. Molar tooth sign in Joubert syndrome: clinical, radiologic, and pathologic significance. J Child Neurol. 1999 Jun;14(6):368-76. doi: 10.1177/088307389901400605. Citation on PubMed
- Parisi M, Glass I. Joubert Syndrome. 2003 Jul 9 [updated 2017 Jun 29]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1325/ Citation on PubMed
- Parisi MA. Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet C Semin Med Genet. 2009 Nov 15;151C(4):326-40. doi: 10.1002/ajmg.c.30229. Citation on PubMed or Free article on PubMed Central
- Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013 Sep;12(9):894-905. doi: 10.1016/S1474-4422(13)70136-4. Epub 2013 Jul 17. Citation on PubMed or Free article on PubMed Central
- Valente EM, Dallapiccola B, Bertini E. Joubert syndrome and related disorders. Handb Clin Neurol. 2013;113:1879-88. doi: 10.1016/B978-0-444-59565-2.00058-7. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.